MPS I (ムコ多糖症 1 型) とは何ですか?
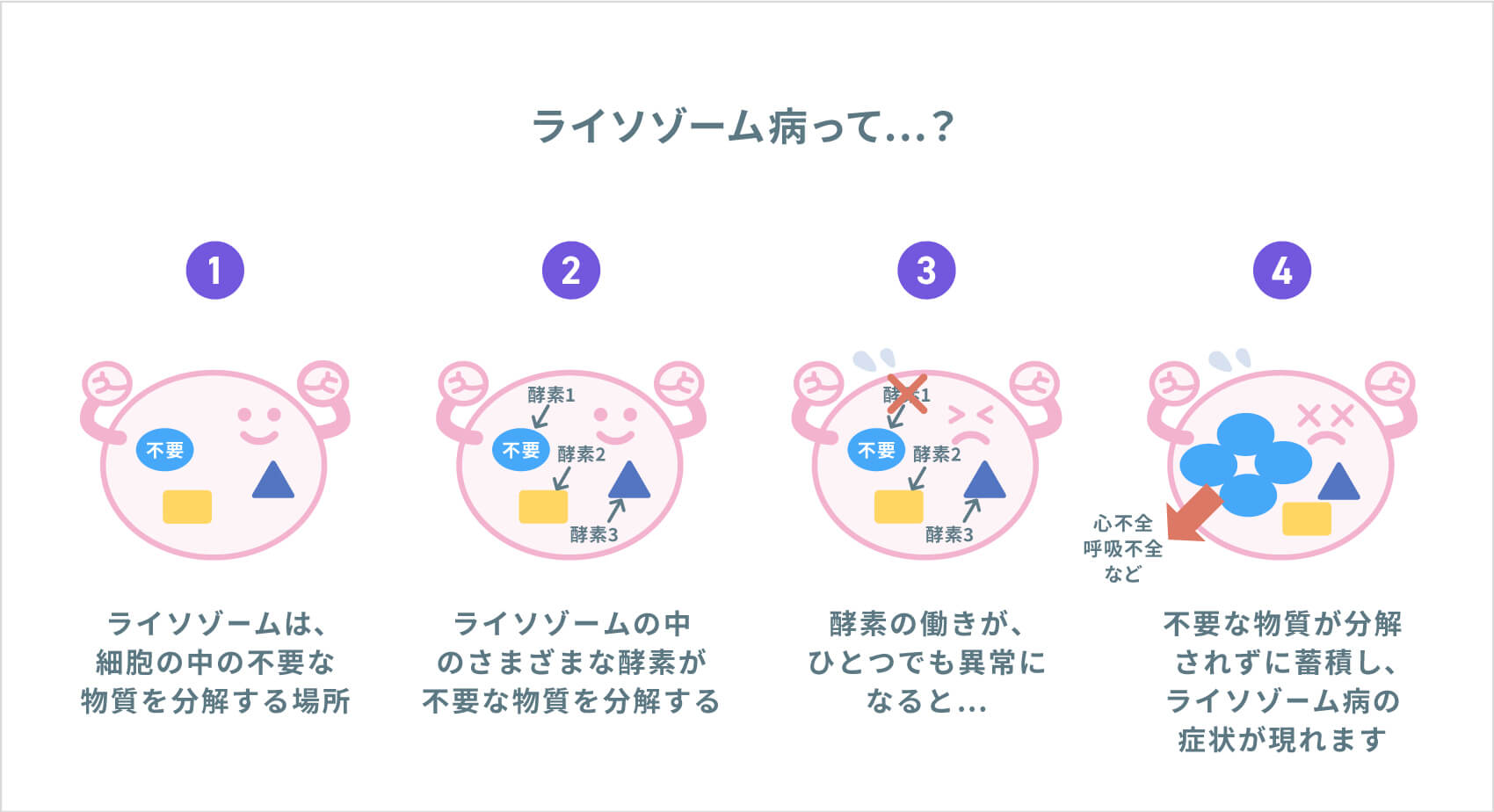
MSP I (ムコ多糖症 1 型) は、リソソーム酵素の欠乏によって引き起こされる最も一般的なタイプの代謝障害です。リソソーム酵素は、多くの異なる種類の物質を分解する役割を果たす細胞小器官である細胞のリソソーム内に含まれています。
MSP I は、第 4 染色体上のアルファ-L-イズロニダーゼをコードする遺伝子の変異によって引き起こされ、結合組織の主要構成要素であるグリコサミノグリカンを分解するために必要なリソソーム酵素が欠損します。
この遺伝子にはさまざまな変異が存在します。さまざまな突然変異は、MPS IH (ハーラー症候群)、MPS IS (シャイエ症候群)、MPS IH/S (ハーラー/シャイエ症候群) などを引き起こします。
MPS IS、MPS IH/S、および MPS IH は軽度の症状 (MPS IS) から最も重篤な症状 (MPS IH) までの範囲の臨床症状であると考えられるため、この記事では一般的に MPS I を単一の症候群として説明します。
MPS I の兆候と症状は何ですか?
前述したように、MPS の兆候と症状はさまざまな重症度を示す場合があります。
MPS I の兆候と症状には次のようなものがあります。
- 顔の特徴の粗大化(通常、最初の異常は生後 3 ~ 6 か月で検出されます)
- 厚い唇で拡大された口
- 頭部拡大
- 慢性的な鼻水
- 進行性の角膜混濁
- 網膜変性
- 脾腫
- 鼠径ヘルニアおよび臍ヘルニア
- 腹部の臓器の肥大
- 骨格異形成は低身長を引き起こす可能性があります
- 関節の硬さ
- 心臓弁膜症、特に大動脈弁膜症
- 頻繁な呼吸器感染症
- 最大機能年齢レベルが約2~4歳に達する発達遅延
最も重度の症状は、MPS IH (ハーラー症候群) で発生します。
MPS I の原因は何ですか?
MPS I の原因は、リソソーム酵素 αL-イズロニダーゼの欠損を引き起こす染色体 4 上の遺伝子変異であり、その結果、組織や器官にムコ多糖が蓄積し、その活性と発達を妨げます。
MPS I はどのように受け継がれるのでしょうか?
MPS I は常染色体劣性遺伝します。その結果、MPS I を患うほぼすべての人は、それぞれの親から劣性遺伝子を受け継いでいます。両親が両方とも劣性遺伝子を持っている場合、子供がそれぞれの親から常染色体劣性遺伝子を受け取り、MPS I を発症する確率は約 4 分の 1 です。
医療専門家は MPS I をどのように治療し管理しますか?
MPS I は、次のような症状を軽減するために医師チーム (前述の専門家を参照) によって治療されます。
MPS I型患者の余命はどれくらいですか?
MPS I の平均余命は非常にばらつきがあります。平均余命は病気の重症度に関連しています。たとえば、最も軽度の MPS I (MPS IS) 型の人は、かなり正常な寿命を持つ可能性がありますが、中間型 (MPS IH/S) の人は、通常、10 代または成人初期まで生きます。重度の MPS I(MPS IH またはハーラー症候群)の患者が 10 年を超えて生きることはほとんどありません。
MPS I のヘルプはどこで見つけられますか?
National MPS Society は、患者とその家族のサポート グループを見つけるのに役立つ追加情報を提供しています (フリーダイヤル番号は 877 MPS 1001)。